
| Molecular progression of SHH-activated medulloblastomas
Medulloblastoma (MB) comprises four main molecular MB subgroups (WNT, SHH, Group 3 and Group 4) with divergent biology, outcomes and subgroup-specific differences in relapses [1, 2, 3, 4, 5, 6]. Metastatic recurrences are most common in Group 3 and 4 MB, where metastases from a single patient are genetically similar to each other, but highly divergent from the corresponding primary tumor [2, 3, 4, 5, 6]. Local recurrences in the tumor bed are more frequent in SHH-activated tumors but progression-associated molecular aberrations for this MB variant remain unclear [2, 6]. To assess a biological evolution of SHH MB, we analyzed molecular changes appearing during local regrowth of desmoplastic nodular MB (DNMB). Eleven pairs of primary and recurrent DNMB samples were analyzed with DNA- and RNA-based methods. Tumor samples were obtained by resection from three infants, two children, and six adults older than 17 years (all at M0 stage; Suppl. Table). After primary resection, infants...
|
| ALK-positive histiocytosis with KIF5B - ALK fusion in the central nervous system
Histiocytic disorders are uncommon and often affect multiple organ systems. They pose diagnostic challenges because of their rarity and the fact that the nosology of these lesions is still being decided. ALK-positive histiocytosis is one of the newest subtypes and was originally described about 10 years ago, wherein there was a predilection for neonates and infants with multi-organ involvement [1]. Since then, ten additional cases have been reported, with only one having exclusive intracranial disease, along with involvement of the cavernous sinus [2, 3]. Here, we report two additional cases with exclusive involvement of the central nervous system.
Case 1 is a 7-year-old girl who presented with a 1-month history of headaches and vomiting. Magnetic resonance imaging (MRI) showed an infiltrating 3 cm mass in the cerebellar vermis. The mass was associated with diffusion restriction and was radiologically suspicious for medulloblastoma (Fig. 1a). She underwent gross total resection...
KeywordsHistiocytic ALK-positive histiocytosis KIF5B-ALK fusion Central nervous system |
| Novel Alzheimer’s disease risk genes: exhaustive investigation is paramount
This year marks the 10th anniversary of the publication of two genome-wide association studies (GWAS) that heralded a change in the field of complex genetics of late-onset Alzheimer’s disease (AD) [1, 2]. Until then, only APOE ɛ4 was unquestionably recognized as a genetic risk factor for AD because of its large effect on disease risk: the so-called low-hanging fruit. These two papers, however, instilled confidence in the ability of the GWAS design to also detect genetic risk loci with smaller effects on AD susceptibility [1, 2]. Since then, a widespread adoption of the GWAS approach has resulted in the identification of many AD risk genes and loci that proved to be consistent across studies. The GWAS data in turn have led to an explosion of subsequent studies of epidemiological or bioinformatic nature, e.g. correlating GWAS-identified SNPs with biomarker phenotypes, or construing information about pathophysiological pathways based on the associated SNPs, or combining alleles at associated SNPs across the genome into polygenic risk profiles. But underneath all this is the realization that GWAS typically provide indirect association signals due to the use of ‘tag’ SNPs. The still undetected genetic variants that directly modulate disease risk at GWAS loci could have a considerably different strength of effect, or uncover molecular mechanisms and pathways involved in AD risk that diverge from current knowledge. This realization has boosted in-depth molecular investigations in genes and loci identified in Alzheimer’s disease GWAS.
The review cluster in this issue of Acta Neuropathologica provides detailed discussions of three of these AD risk genes (SORL1, CD33 and ABCA7) for which significant advances have been made in identifying genetic variants that directly modulate Alzheimer’s disease risk [3, 4, 5]. A fourth paper gives a bird’s-eye view of the new genetic landscape of AD, and uses recent experimental evidence on AD genes to give a different interpretation of the GWAS data in the shape of a new model of AD pathogenesis [6].
Several important messages can be gathered from these reviews. Both SORL1 and ABCA7, which were reported as Alzheimer’s disease risk genes based on the association of common variants, were subsequently shown to harbor numerous rare deleterious variants which were detected more often in AD patients than in cognitively healthy individuals [3, 5]. While logical in hindsight, these were important breakthroughs stirring the prevailing notion that the genetic architecture of complex AD was dominated by APOE ɛ4 and common variants with small effects. The protective role of the sorLA protein in AD had already been studied extensively, but the functionality of some of its protein domains was still incompletely understood [3]. The effect ABCA7 may exert on AD risk is still uncertain [5]. The rare predicted loss-of-function variants in ABCA7 and SORL1 can now guide the design of experimental investigation of their molecular effect in the context of AD.
The multitude of different rare variants in SORL1 and ABCA7, combined with the strength of their risk increasing effect, means that their impact at the population level is not negligible. In their review, Campion and colleagues report that 3.6% of all AD patients and up to 4.8% of early onset AD patients carry a protein truncating mutation or rare predicted pathogenic missense mutation in SORL1 [3]. Another 4.4% of European AD patients carry a rare ABCA7protein truncating mutation (overlap between SORL1 and ABCA7 carriers assumed to be minimal) [5]. Odds ratios well exceed those of common variant associations in GWAS. This begs the question whether current polygenic risk profiling efforts (for scientific purpose) would not benefit from incorporating information on carrier status of rare risk variants. Campion and colleagues do caution against current implementation of genetic screening for these variants in a clinical setting, in the absence of functional and segregation data [3]. This is clearly exemplified by the protein truncating variants in ABCA7 [5]. At first glance these protein truncating variants are considered to lead to 50% loss of protein due nonsense mediated mRNA decay, but cDNA sequencing of several different ABCA7 variants revealed both escape from nonsense mediated mRNA decay and unknown splicing events that could restore the reading frame. The extent to which this occurs differs from carrier to carrier, troubling straightforward interpretation of the impact of such variants on individual disease risk.
While neither rare variants in SORL1 nor in ABCA7 can account for the GWAS association signals at these loci, common functional polymorphisms explaining the GWAS signals in ABCA7 as well as in CD33 have been identified, as reviewed [4, 5]. In ABCA7, a common protein truncating variant caused by a large deletion explains the GWAS association signal in African Americans, and a pathogenic repeat expansion explains the GWAS association signal in Europeans (reviewed in Ref. [5]). Corroborated by both earlier and more recent evidence in different loci [7, 8], these findings illustrate that the repertoire of genetic variation to be investigated as risk factors for AD should be broadened. Technologies such as long-read sequencing will facilitate this in the years to come.
Intriguingly, while CD33 was among the first GWAS-identified genes in which a functional risk variant was detected, its association with AD in GWAS is not always replicated. The functional polymorphism affects CD33 exon 2 splicing efficiency. The AD-protective allele results in an increase of an isoform lacking exon 2, which encodes the IgV domain involved in ligand binding. This observation has led to the formulation of a loss of function hypothesis for CD33 in AD risk. In their review, however, Estus and colleagues propose a new gain of function hypothesis based on recent experimental evidence [4]. While additional evidence will be required to reconcile all existing data in this new model, progress on CD33 exemplifies that in silico pathway-based analyses that lack this level of detail are unlikely to accurately approximate the pathogenesis of AD.
A similar message can be derived from the review of Dourlen et al. [6]. In their timely synopsis of the genetic landscape of AD, they first discuss the new AD risk genes in relation to the amyloid cascade hypothesis, including genes involved in APP metabolism and Aß peptide production, Aß peptide degradation and clearance, and Aß peptide toxicity, as well as tau toxicity. Evidence from high-throughput molecular approaches on novel AD genes, however, motivated Dourlen et al. to propose a new paradigm for AD pathogenesis in which they assign a central role to the focal adhesion pathway and dysregulation of synaptic plasticity. As they point out in their review, this pathway would not have been identified when relying on in silico analyses of GWAS gene lists alone [6].
Ideally, this new model will spark debate and stimulate efforts to verify or refute it, which in turn will yield further insights. An attractive feature of the model is its circular nature and the notion behind it that individual patients may have different entry points into the vicious cycle, requiring personally adapted treatment strategies. The next challenge will be to delineate identifiable subgroups of individuals that are likely to share an entry point into the vicious cycle of AD pathology. Genetically defined subtypes could be a promising start.
|
| Lack of H3K27 trimethylation is associated with 1p/19q codeletion in diffuse gliomas
The current World Health Organisation (WHO) Classification of Central Nervous System Tumours defines oligodendrogliomas by IDH mutation and 1p/19q codeletion [12]. Oligodendrogliomas differ from diffuse astrocytomas regarding genetic alterations in telomere maintenance mechanisms by frequently displaying TERT promoter mutations while astrocytomas typically exhibit ATRX (a-thalassaemia/mental retardation syndrome X-linked) mutations leading to alternative lengthening of telomeres (ALT) [1, 2, 7, 10, 13]. The distinction between both glioma types is crucial since it has a considerable impact on both patient treatment and outcome.
Currently, methodological guidelines for the assessment of 1p/19q codeletion are missing and commonly PCR-based loss of heterozygosity analyses or fluorescent in situ hybridization (FISH) are used. IDH mutant astrocytomas and oligodendrogliomas can also be distinguished by DNA methylation-based profiling, which allows 1p/19q assessment via calculated copy number profiles (CNP) [4]. Infinium 850 k EPIC array is a highly reliable and accurate technique for DNA methylation analysis but can currently only be performed by specialized laboratories as it requires substantial investment in infrastructure and personnel.
Trimethylation at lysine 27 of histone 3 (H3K27me3) is a repressive histone mark associated with inhibition of transcription and represents a post-translational modification set by EZH2, a component of the Polycomb repressive complex 2 [5, 15, 16]. In a paediatric ependymoma cohort, reduction of H3K27me3 defines a subgroup of posterior fossa ependymomas with poor prognosis [3, 17]. Likewise, WHO grade I and II meningiomas display a higher risk of recurrence when H3K27me3 is lost [9]. However, since comprehensive data about H3K27me3 in IDH mutant gliomas is missing, we screened an epigenetically well-defined glioma cohort consisting of 26 IDH mutant and 1p/19q codeleted oligodendrogliomas, 34 IDH mutant astrocytomas and 101 IDH wildtype glioblastomas for potential differences in H3K27 trimethylation on protein level.
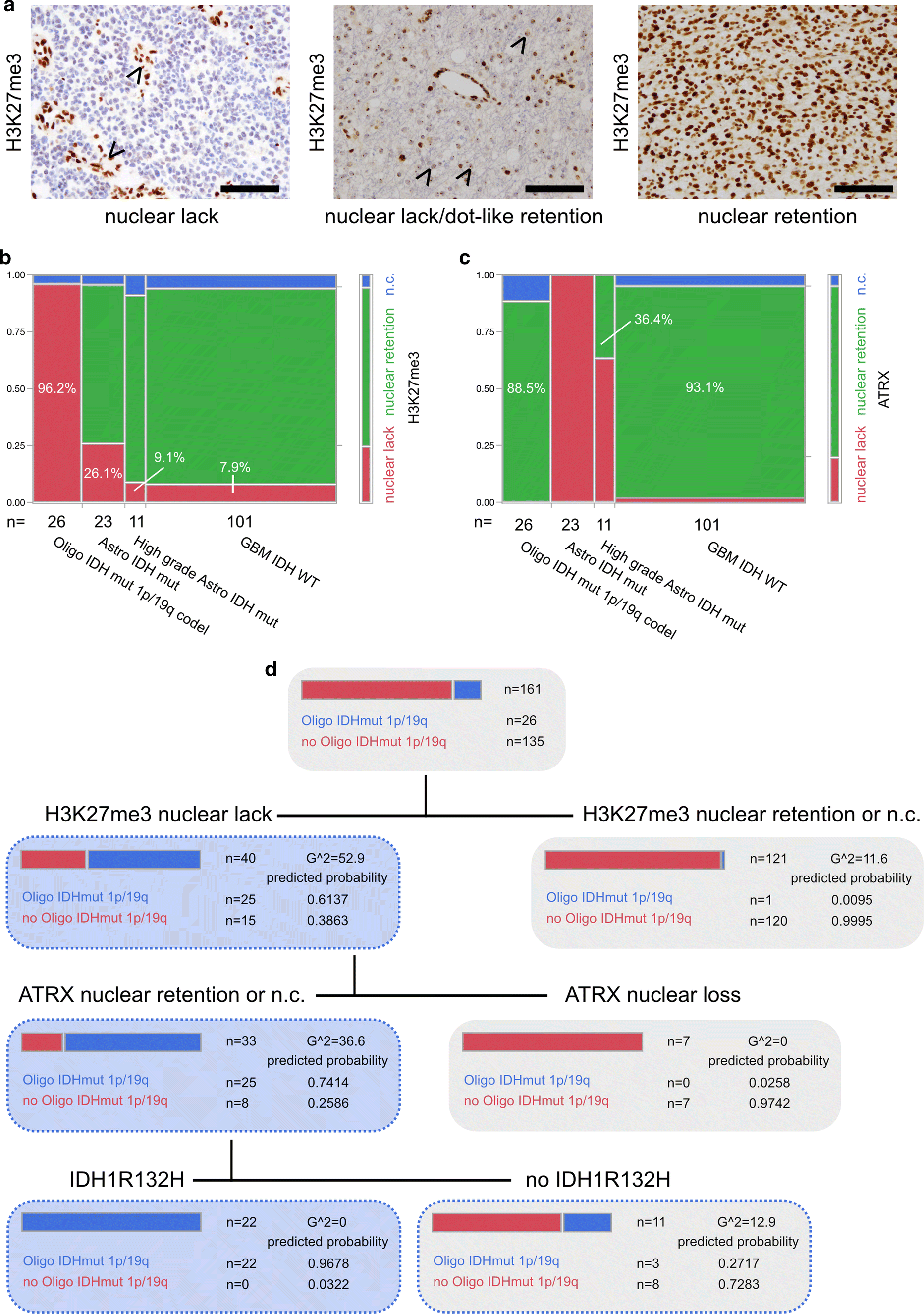
Diffuse gliomas showed differences in H3K27me3 staining by either displaying nuclear retention or lack of H3K27me3 immunoreactivity (Fig. 1a). In line with H3K27me3 functioning as a transcriptional silencing mechanism via chromatin remodelling for X-inactivation, we also noticed nuclear lack of H3K27me3 in combination with preserved dot-like staining of the inactivated X chromosome in a subgroup of female glioma patients (Fig. 1a). Furthermore, H3K27me3 turned out to be an indicator of patient survival: patients with lack of H3K27me3 expression showed a significantly better prognosis than patients with retained H3K27me3 staining (Supplementary Fig. 1).
Fig. 1
a Immunohistochemistry against H3K27me3. Arrowheads pointing at retained nuclear staining in endothelial cells while tumour cells show a lack of staining (IDH mutant and 1p/19q codeleted oligodendroglioma, left panel). Arrowheads pointing at dot-like H3K27me3 retention in otherwise H3K27me3-negative tumour cells in a case of a female patient (IDH mutant and 1p/19q codeleted oligodendroglioma, middle panel). Nuclear retention of H3K27me3 in a glioblastoma IDH-WT specimen (right panel) (all scale bars 100 µm). b H3K27me3 and c ATRX expression in epigenetically defined glioma subclasses [4] (n.c. non-conclusive). d Decision tree of recursive partitioning using DNA methylation-based classifier diagnosis “IDH mutant and 1p/19q codeleted oligodendroglioma” as dependent variable and H3K27me3, ATRX and IDH1R132H staining as predictors. Decision tree showing results of best split
Next, we set out to compare H3K27me3 immunoreactivity with DNA methylation classes. None of the investigated tumours showed a histone 3 lysine 27 M mutation, as assessed by a mutation-specific antibody (Supplementary Fig. 1). Most interestingly, in IDH mutant and 1p/19q codeleted oligodendroglioma, H3K27me3 retention was never observed, the abundant number of cases (25/26) showed clear lack of nuclear H3K27me3 staining and only one case had to be deemed non-conclusive (Fig. 1b). Gliomas belonging to the DNA methylation class of IDH mutant astrocytomas or IDH mutant high-grade astrocytomas were characterized by nuclear H3K27me3 retention in 69.6% and 81.8%, respectively (Fig. 1b). Malignant gliomas of the DNA methylation class of IDH wildtype glioblastoma predominantly presented with preserved nuclear H3K27me3 staining (86.1%, Fig. 1b). Interestingly, in IDH wildtype glioblastomas, lack of nuclear staining was exclusively associated with methylation subclasses “mesenchymal” (3 out of 36) and “RTK I” (5 out of 26) (Supplementary Fig. 1c displaying examples of H3K27me3 staining in different glioma subtypes). As loss of nuclear ATRX staining and 1p/19q codeletion are mostly mutually exclusive, ATRX immunohistochemistry is useful in discrimination between an oligodendroglial and astrocytic tumour lineage [6, 8, 11, 18]. Nevertheless, non-conclusive ATRX staining impeded diagnosis of oligodendroglioma in 11.5% and might have been misleading in up to 36.4% of IDH mutant high-grade astrocytomas which presented with retained nuclear ATRX staining (Fig. 1c).
To assess the predictive value of H3K27me3 expression in diffuse gliomas we deployed a recursive partitioning model for the prediction of the DNA methylation class IDH mutant and 1p/19q codeleted oligodendroglioma with H3K27me3, ATRX and IDH1R132H staining as predictors (Fig. 1d). Sequential immunohistochemistry against the aforementioned antigens revealed that diffuse gliomas with lack of nuclear H3K27me3 staining, retention or non-conclusive nuclear ATRX staining and IDH1R132H mutation are 1p/19q codeleted oligodendrogliomas with a predicted probability of 0.9678 (Fig. 1d, see also results of a validation cohort in Supplementary Fig. 2).
Our results point to differences in histone H3K27 modification in diffuse glial tumours which are advantageous for the clinically relevant discrimination between astrocytic and oligodendroglial tumour lineage. Results of our prediction model suggests the usage of an antibody panel including antibodies against IDH1R132H, H3K27M (exclusion of mutation), ATRX and H3K27me3 for the prediction of the DNA methylation class “oligodendroglioma, IDH mutant and 1p/19q codeleted” (Supplementary Table 1). In diffuse gliomas harbouring a lack of nuclear H3K27me3 in addition to retained or non-conclusive ATRX staining but no IDH1R132H-mutation, IDH sequencing should be followed up. Small tumour biopsies or infiltration zones require careful evaluation of immunostainings (Supplementary Fig. 1c).
With regard to tumour biology, the lack of nuclear H3K27me3 in oligodendrogliomas is surprising given the fact that increasing levels of 2-hydroxyglutarate in cells harbouring an IDH mutation are known to impair demethylation of other repressive histone marks, such as H3K9me3, with resulting gain of histone methylation [14]. The resulting chromatin compaction would promote CpG island hypermethylation phenotype [19]. The relation between 1p/19q codeletion and global lack of H3K27me3 requires further investigation.
Supplementary material
401_2019_2025_MOESM1_ESM.tiff (8.4 mb)
Supplementary Figure 1: (a) Kaplan-Meier survival analyses of a non-molecularly classified tissue micro array (TMA) cohort (1998-2011) of different diffuse gliomas including oligodendrogliomas, astrocytomas, and glioblastomas. (b) Results of IDH1R132H, ATRX, H3K27me3 and H3K27M immunohistochemistry in epigenetically defined glioma subclasses. (c) Collection of H3K27me3 staining patterns in different glioma subtypes (TIFF 8558 kb)
401_2019_2025_MOESM2_ESM.tiff (1.5 mb)
Supplementary Figure 2: Immunohistochemical analyses for IDH1R132H, ATRX, H3K27me3 and H3K27M in the epigenetically classified validation cohort consisting of 18 IDH-mutant gliomas (1p/19q codeleted oligodendrogliomas (n=9); astrocytomas (n=9)) (TIFF 1556 kb)
401_2019_2025_MOESM3_ESM.docx (12 kb)
Supplementary Table 1: Antibodies, supplier and dilution for immunohistochemistry (DOCX 12 kb)
|
| Neurotoxicology: an update on epidemiology, mechanisms, and pathology
The adverse impact of environmental chemicals on the human brain has been recognized as an important endpoint in toxicology. In one of the most influential toxicology textbooks, the authors stated in their introduction that … “the target organ of toxicity most frequently involved in systemic toxicity is the CNS” [1]. Accordingly, the 2nd edition of “Experimental and clinical neurotoxicology”, edited by Peter Spencer and Herbert H. Schaumburg [2], listed more than 450 compounds that are suspected or proven neurotoxins/toxicants in humans. Naturally occurring neurotoxins, such as domoic acid (DA) or tetrodotoxin (TTX), are among the most potent poisons that can be found in nature, whereas organic solvents are among the man-made neurotoxicants shown to be associated with severe damage in the central and peripheral nervous system [3]. Already in the early 1980s, Acta Neuropathologica published human biopsy pictures showing demyelination of giant axons taken from the terminal portion of the musculocutaneous nerve of the leg of a patient chronically exposed to n-hexane and methylethylketone [4]. Later, details about specific neuropathological changes after chronic solvent abuse via inhalation could be shown in 88 autopsy cases [5]. In addition to macroscopic findings, such as enlarged ventricles, white matter abnormalities, and cerebral atrophy, the study showed that chronic solvent leukoencephalopathy can be identified by birefringent PAS-staining macrophages and reactive microglia in the white matter. However, the study was not able to disentangle the neuropathological effects of the different solvents that the cases abused simultaneously. Nevertheless, such a neuropathological differential diagnosis was possible in the field of aluminum (Al) neurotoxicity. The neurotoxicity of Al is well known and described in detail for various endpoints and species [6]. Al exposure has also been linked to the pathogenesis of Alzheimer’s Disease (AD) [7]. Aluminum neurotoxicity is also thought to play a role in dialysis-associated encephalopathy (DAE) where Al-containing drugs are used to control hyperphosphatemia, and dialysis dementia has been frequently observed as clinical outcome. Despite the proven neurotoxicity of Al, another well-conducted neuropathology study by Reusche et al. [8] could show that the changes in human brain tissue in DAE patients differed markedly from AD patients. DAE patients did not show AD-type neurofibrillary tangles (NFT) above the normal or expected age-related changes, even though the Al concentrations in the brain samples were markedly increased. These examples illustrate the valuable contributions of neuropathology to the area of toxicology, in particular neurotoxicology. When searching the electronic archive of Acta Neuropathologica, one can find approximately 120 publications related to neurotoxicology but only a few, like the examples given before, included histopathological analyses in human brain tissue. Moreover, these studies were mostly performed under conditions of high exposures or even intoxication. During the last decades, only a few neurotoxicity studies or reviews have been published that have a strong focus on human neuropathology (e.g., [9]). This deficit has been identified by the former Editor-in-Chief, Werner Paulus, who developed and initiated the idea of a cluster of reviews addressing current hot topics in neurotoxicology. This was a challenging endeavour as there are some differences between these obviously-related disciplines.
Why the disparity between these two disciplines within neuroscience?
There are some reasons for this weak association between neuropathology and toxicology that are inherent to the scientific and societal aims of toxicology. Toxicology tries to contribute to risk assessment procedures, and consequently, studies are needed that determine dose–response relationships between the magnitude of exposure and the probability to cause adverse health effects. While the histopathological examination of other target organs (e.g., liver and lung) is still a cornerstone in toxicological guideline studies (e.g., as proposed by the Organization for Economic Cooperation and Development (OECD) [10]), neurotoxicity testing in rodents also relies on functional or behavioral testing (e.g., motor activity [11]). With respect to animal studies, the various regulatory agencies also provide some guidance regarding neuropathology assessment [12], but these examinations are more often a rough estimate of neuropathological changes not fully exploiting the current state of the art in neuropathology. Accordingly, behavioral tests in animals, which have been suggested as an important, sensitive, and apical endpoint in chemical risk assessment, are used more frequently [13, 14], and “safe” levels of exposure are often derived from these endpoints, as in the case of the solvent toluene [15] that also causes neuropathological effects at higher doses [5]. Recently, and related to the efforts of developing alternative methods in toxicity testing [16], behavioral testing in zebrafish has been proposed as a model to investigate adverse outcomes in a whole organism upon exposure to neurotoxic compounds [17]. Accordingly, neuropathology is often only a minor point in neurotoxicity testing, both in guideline studies and scientific research.
In humans, epidemiological studies of exposed populations (e.g., occupationally or environmentally) are the only source to derive dose–response relationships, and here, the availability of brain tissue is limited. Even in cases like the solvent abusers [5], it is difficult to link individual exposure data to these brain samples. Therefore, in human studies, neurobehavioral testing has been the only or at least most important endpoint for the assessment of adverse health effects to the human brain [18]. In addition to the strong focus on behavioral measures as a “surrogate marker” of impaired brain functions, the deficit of neurophysiological and even pathological human data in neurotoxicology was also caused by (a) the lack of non-invasive methods to investigate brain functions in vivo, and (b) the limited availability of neuroimaging techniques, such as functional and structural Magnetic Resonance Imaging (MRI). Various neurophysiological techniques have become increasingly available in epidemiological studies among workers, e.g., manganese-exposed welders [19], and thereby, the knowledge about neurotoxic mechanisms can be used for the selection of sensitive, neurophysiological endpoints in experimental or epidemiological studies [20, 21]. Later, the validation of such neuroimaging findings in brain tissue could provide conclusive information about dose-dependent neuropathological changes after neurotoxic exposures. Thereby, the impact of neurotoxic exposures could also be evaluated more precisely as these findings can be compared to the neuropathology of aging [22] or neurogenerative diseases [23].
Recent developments that might facilitate interactions between neuropathology and toxicology
The ongoing paradigm shift towards toxicological testing strategies that are based on mechanistic knowledge about the perturbations of molecular and cellular events within neural cells and networks [24, 25] might promote or revive the integration of neuropathology into toxicology. One conceptional tool that is relevant here is the idea of an “Adverse Outcome Pathway” [26] that has be adopted by neurotoxicity [27]. Here, the molecular initiating event (MIE), e.g., the chronic antagonism of N-methyl-d-aspartate receptors (NMDARs) during brain development, should cause impairment of learning and memory abilities in children. The apical endpoint of this adverse effect might be the reduced IQ of children as shown for low-level lead exposures [28]. Such adverse effects of environmental toxicants have also been addressed in neurophysiological and pathological studies, as summarized for prenatal exposure to maternal cigarette smoking (PEMCS) [29]. In particular, the MRI readouts were able to detect adverse neurotoxic effects in exposed children, such as thinner orbitofrontal, middle frontal, and parahippocampal cortices in smoke-exposed children [30]. These morphometric readouts were more sensitive than the Wechsler Intelligence Scale for Children. Moreover, in this review, neuropathological findings from animal studies were able to provide more details about the pathological changes in the brain of rats (e.g., increased spine density in the granule cells, and terminal and basal dendrites of the pyramidal neurons of CA3 and CA1 of the hippocampus). These examples clearly showed that the paradigm change in neurotoxicology might be a chance to intensify the collaboration between neuropathology and neurotoxicology.
The present neurotoxicity review cluster is intended to be a first step in this direction and might encourage researchers from both disciplines to intensify collaborations. Two hot topics from neurotoxicology, namely the exposure to pesticides [19] and polychlorinated biphenyls (PCBs) [31], will be addressed in detail. Both reviews will provide some information about the history of these neurotoxicants, their diversity with respect to chemistry, and their ability to persist in the environment and human tissue even for decades. Summaries of in vivo and in vitro studies describing the different neurotoxic mechanisms that have been discovered are presented, and their relevance for the epidemiological findings has been discussed. While the review on PBC has a stronger focus on developmental neurotoxicity, the pesticide review will also address possible associations of this class of neurotoxicants with neurodegenerative diseases. Finally, both reviews focus on the need for a comprehensive characterization of the neurotoxic properties of new chemicals that may be developed to substitute the two groups of chemicals reviewed in this cluster.
I am confident that this cluster of two reviews is an excellent starting point to stimulate the dialogue between these two “neuro” research areas.
|
RhoA regulates translation of the Nogo-A decoy SPARC in white matter-invading glioblastomasAbstract
Glioblastomas strongly invade the brain by infiltrating into the white matter along myelinated nerve fiber tracts even though the myelin protein Nogo-A prevents cell migration by activating inhibitory RhoA signaling. The mechanisms behind this long-known phenomenon remained elusive so far, precluding a targeted therapeutic intervention. This study demonstrates that the prevalent activation of AKT in gliomas increases the ER protein-folding capacity and enables tumor cells to utilize a side effect of RhoA activation: the perturbation of the IRE1α-mediated decay of SPARC mRNA. Once translation is initiated, glioblastoma cells rapidly secrete SPARC to block Nogo-A from inhibiting migration via RhoA. By advanced ultramicroscopy for studying single-cell invasion in whole, undissected mouse brains, we show that gliomas require SPARC for invading into white matter structures. SPARC depletion reduces tumor dissemination that significantly prolongs survival and improves response to cytostatic therapy. Our finding of a novel RhoA-IRE1 axis provides a druggable target for interfering with SPARC production and underscores its therapeutic value.
|
A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevityAbstract
The genetic variant rs72824905-G (minor allele) in the PLCG2 gene was previously associated with a reduced Alzheimer’s disease risk (AD). The role of PLCG2 in immune system signaling suggests it may also protect against other neurodegenerative diseases and possibly associates with longevity. We studied the effect of the rs72824905-G on seven neurodegenerative diseases and longevity, using 53,627 patients, 3,516 long-lived individuals and 149,290 study-matched controls. We replicated the association of rs72824905-G with reduced AD risk and we found an association with reduced risk of dementia with Lewy bodies (DLB) and frontotemporal dementia (FTD). We did not find evidence for an effect on Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) risks, despite adequate sample sizes. Conversely, the rs72824905-G allele was associated with increased likelihood of longevity. By-proxy analyses in the UK Biobank supported the associations with both dementia and longevity. Concluding, rs72824905-G has a protective effect against multiple neurodegenerative diseases indicating shared aspects of disease etiology. Our findings merit studying the PLCγ2 pathway as drug-target.
|
Evaluation of CD33 as a genetic risk factor for Alzheimer’s diseaseAbstract
In 2011, genome-wide association studies implicated a polymorphism near CD33 as a genetic risk factor for Alzheimer’s disease. This finding sparked interest in this member of the sialic acid-binding immunoglobulin-type lectin family which is linked to innate immunity. Subsequent studies found that CD33 is expressed in microglia in the brain and then investigated the molecular mechanism underlying the CD33 genetic association with Alzheimer’s disease. The allele that protects from Alzheimer’s disease acts predominately to increase a CD33 isoform lacking exon 2 at the expense of the prototypic, full-length CD33 that contains exon 2. Since this exon encodes the sialic acid ligand-binding domain, the finding that the loss of exon 2 was associated with decreased Alzheimer’s disease risk was interpreted as meaning that a decrease in functional CD33 and its associated immune suppression was protective from Alzheimer’s disease. However, this interpretation may need to be reconsidered given current findings that a genetic deletion which abrogates CD33 is not associated with Alzheimer’s disease risk. Therefore, integrating currently available findings leads us to propose a model wherein the CD33 isoform lacking the ligand-binding domain represents a gain of function variant that reduces Alzheimer’s disease risk.
|
Mutational patterns and regulatory networks in epigenetic subgroups of meningiomaAbstract
DNA methylation patterns delineate clinically relevant subgroups of meningioma. We previously established the six meningioma methylation classes (MC) benign 1–3, intermediate A and B, and malignant. Here, we set out to identify subgroup-specific mutational patterns and gene regulation. Whole genome sequencing was performed on 62 samples across all MCs and WHO grades from 62 patients with matched blood control, including 40 sporadic meningiomas and 22 meningiomas arising after radiation (Mrad). RNA sequencing was added for 18 of these cases and chromatin-immunoprecipitation for histone H3 lysine 27 acetylation (H3K27ac) followed by sequencing (ChIP-seq) for 16 samples. Besides the known mutations in meningioma, structural variants were found as the mechanism of NF2 inactivation in a small subset (5%) of sporadic meningiomas, similar to previous reports for Mrad. Aberrations of DMD were found to be enriched in MCs with NF2 mutations, and DMD was among the most differentially upregulated genes in NF2 mutant compared to NF2 wild-type cases. The mutational signature AC3, which has been associated with defects in homologous recombination repair (HRR), was detected in both sporadic meningioma and Mrad, but widely distributed across the genome in sporadic cases and enriched near genomic breakpoints in Mrad. Compared to the other MCs, the number of single nucleotide variants matching the AC3 pattern was significantly higher in the malignant MC, which also exhibited higher genomic instability, determined by the numbers of both large segments affected by copy number alterations and breakpoints between large segments. ChIP-seq analysis for H3K27ac revealed a specific activation of genes regulated by the transcription factor FOXM1 in the malignant MC. This analysis also revealed a super enhancer near the HOXD gene cluster in this MC, which, together with general upregulation of HOX genes in the malignant MC, indicates a role of HOX genes in meningioma aggressiveness. This data elucidates the biological mechanisms rendering different epigenetic subgroups of meningiomas, and suggests leveraging HRR as a novel therapeutic target.
|
SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing dataAbstract
Massive parallel sequencing recently allowed the identification of three genes carrying a higher burden of rare, protein-truncating and missense predicted damaging variants in Alzheimer disease (AD) cases as compared to controls: TREM2, SORL1, and ABCA7. SORL1 encodes SorLA, a key protein involved in the processing of the amyloid-beta (Aβ) precursor protein (APP) and the secretion of the Aβ peptide, the aggregation of which triggers AD pathophysiology. Common SORL1 single nucleotide polymorphisms had originally been associated with AD with modest odds ratios (ORs). The association of AD with rare SORL1 coding variants has been demonstrated at the gene level by aggregating protein-truncating (PTV) and rare predicted damaging missense variants. In addition to the loss of SorLA function induced by PTVs, a few missense variants were studied in vitro, showing diverse degrees of decreased SorLA function and leading to increased Aβ secretion. However, the exact functional consequences of most of the missense variants remain to be determined as well as corresponding levels of AD risk. Hereby we review the evidence of the association of SORL1 common and rare variants with AD risk and conduct a meta-analysis of published data on SORL1 rare variants in five large sequencing studies. We observe a significant enrichment in PTVs with ORs of 12.29 (95% confidence interval = [4.22–35.78]) among all AD cases and 27.50 [7.38–102.42] among early-onset cases. Rare [minor allele frequency (MAF) < 1%] and ultra-rare (MAF < 10−4) missense variants that are predicted damaging by 3/3 bioinformatics tools also show significant associations with corresponding ORs of 1.87 [1.54–2.28] and 3.14 [2.30–4.28], respectively. Per-domain analyses show significant association with the APP-binding CR cluster class A repeats and the Aβ-binding VPS10P domains, as well as the fibronectin type III domain, the function of which remains to be specified. These results further support a critical role for SORL1 rare coding variants in AD, although functional and segregation analyses are required to allow an accurate use in a clinical setting.
|
Πληροφορίες
Πέμπτη 1 Αυγούστου 2019
Εγγραφή σε:
Σχόλια ανάρτησης (Atom)
Δεν υπάρχουν σχόλια:
Δημοσίευση σχολίου